Author: Rodrigo Arrangoiz MS, MD, FACS, FSSO
My name is Rodrigo Arrangoiz I am a breast surgeon/ thyroid surgeon / parathyroid surgeon / head and neck surgeon / surgical oncologist that works at Center for Advanced Surgical Oncology in Miami, Florida.
I was trained as a surgeon at Michigan State University from (2005 to 2010) where I was a chief resident in 2010. My surgical oncology and head and neck training was performed at the Fox Chase Cancer Center in Philadelphia from 2010 to 2012. At the same time I underwent a masters in science (Clinical research for health professionals) at the University of Drexel. Through the International Federation of Head and Neck Societies / Memorial Sloan Kettering Cancer Center I performed a two year head and neck surgery and oncology / endocrine fellowship that ended in 2016.
Mi nombre es Rodrigo Arrangoiz, soy cirujano oncólogo / cirujano de tumores de cabeza y cuello / cirujano endocrino que trabaja Center for Advanced Surgical Oncology en Miami, Florida.
Fui entrenado como cirujano en Michigan State University (2005 a 2010 ) donde fui jefe de residentes en 2010. Mi formación en oncología quirúrgica y e n tumores de cabeza y cuello se realizó en el Fox Chase Cancer Center en Filadelfia de 2010 a 2012. Al mismo tiempo, me sometí a una maestría en ciencias (investigación clínica para profesionales de la salud) en la Universidad de Drexel. A través de la Federación Internacional de Sociedades de Cabeza y Cuello / Memorial Sloan Kettering Cancer Center realicé una sub especialidad en cirugía de cabeza y cuello / cirugia endocrina de dos años que terminó en 2016.




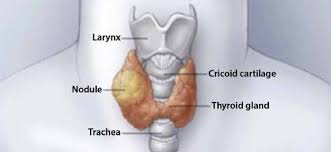
Valoración Inicial de los Nódulos Tiroideos
INTRODUCCIÓN
Los nódulos tiroideos son un problema importante de salud. La prevalencia de los nódulos tiroideos palpables en la población general es de aproximadamente del 5% en mujeres y del 1% en hombres que viven en partes del mundo con suficiente yodo. Por el contrario, el ultrasonido de cuello y tiroides de alta resolución puede detectar nódulos tiroideos en aproximadamente el 19% al 68% de las personas seleccionadas al azar, con frecuencias más altas en las mujeres y en las personas de edad avanzada. La importancia clínica de los nódulos tiroideos reside en la necesidad de descartar el cáncer de tiroides, que ocurre entre el 7% al 15% de los casos, variando según el sexo, la edad, el historial de exposición a la radiación, los antecedentes familiares, entre otros factores.
La detección y el diagnóstico del cáncer de tiroides diferenciado ha evolucionado a lo largo de los años con una mayor utilización del ultrasonido de cuello y tiroides de alta resolución, la biopsia por aspiración con aguja fina (BAAF), las pruebas moleculares, y la tiroglobulina como marcador tumoral sérico. En este capitulo abordaremos el manejo actual basado en evidencia de los nódulos tiroideos.
VALORACIÓN INICIAL DEL NÓDULO TIROIDEO
Al descubrir un nódulo tiroideo, se debe realizar una historia clínica completa y un examen físico centrado en la glándula tiroides y los ganglios linfáticos regionales adyacentes. Los factores históricos pertinentes que pronostican malignidad en un nódulo tiroideo incluyen un historial de radioterapia infantil a la región de la cabeza y cuello, radiación corporal total por trasplante de médula ósea, exposición a radiación ionizante en la infancia o en la adolescencia, carcinoma tiroideo familiar, o síndrome hereditario de cáncer de tiroides (síndrome Cowden, adenomatosis familiar polipoidea, complejo de Carney, síndrome de Werner, neoplasia endocrina múltiple 2A, o neoplasia endocrina múltiple 2B), un nódulo tiroideo con crecimiento rápido y / o ronquera.
Los hallazgos físicos pertinentes que sugieren de una posible malignidad incluyen la parálisis de las cuerdas vocales, la linfadenopatía cervical, y la fijación del nódulo tiroideo a los tejidos circundantes. Con el descubrimiento de un nódulo tiroideo mayor a 1 cm en cualquier diámetro, se debe obtener un nivel sérico de la hormona estimulante de tiroides (TSH) (recomendación 2 American Thyroid Association [ATA]). Si la TSH esta baja, se debe realizar una gammagrafía tiroidea (única indicación hoy en día para realizar este estudio) para documentar si el nódulo tiroideo está hiperfuncionante (“caliente”, es decir, la captación del marcador es mayor que la tiroides normal), isofuncionante (“cálido”, es decir, , la captación del marcador es igual a la tiroides circundante) o no funcionante (“frío”, es decir, tiene una captación menor que el tejido tiroideo). Dado que los nódulos tiroideos hiperfuncionantes rara vez contienen malignidad, si se encuentra uno que corresponde al nódulo en cuestión, no es necesaria una evaluación citológica. Si hay hipertiroidismo evidente o subclínico, se requiere una evaluación adicional con una gammagrafía tiroidea. Niveles séricos altos de TSH, incluso dentro de rangos altos de normalidad, se asocia con un mayor riesgo de malignidad en el nódulo tiroideo, así como un estadio más avanzado del cáncer tiroideo.
Durante la valoración inicial de los nódulos tiroideos no se recomienda obtener de manera rutinaria la tiroglobulina sérica (Tg) (recomendación 3 de la ATA). Los niveles séricos de la Tg pueden estar elevados en la gran mayoría de las enfermedades tiroideas (benignas y malignas) y es una prueba insensible e inespecífica para el cáncer de tiroides. La utilidad de la calcitonina sérica en la valoración inicial de los nódulos tiroides se ha evaluado en estudios prospectivos no aleatorios, con resultados mixtos, por lo tanto, la ATA no puede recomendar ni a favor ni en contra de la medición rutinaria de la calcitonina sérica en pacientes con nódulos tiroideos (recomendación 4 de la ATA).
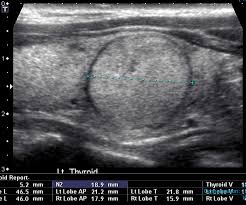
Se debe realizar un ultrasonido de cuello y tiroides de alta resolución en todos los pacientes con sospecha de tener nódulos tiroideos, bocio nodular, o cualquier alteración radiográfica que sugiera un nódulo tiroideo detectado de manera incidental en otro estudio de imagen (tomografía computarizada o resonancia magnética o 18FDG-PET) (recomendación 6 de la ATA). El ultrasonido de cuello y tiroides debe evaluar las siguientes características:
• El parénquima tiroideo (si esta homogéneo o heterogéneo)
• El tamaño de la glándula tiroides
• El tamaño, ubicación, y características ultrasonográficas de cualquier nódulo
• La presencia o ausencia de ganglios linfáticos cervicales sospechosos en los compartimentos centrales o laterales.
Las características que se deben valorar en el ultrasonido son:
• Tamaño del nódulo (en tres dimensiones)
• La ubicación (ejemplo – lóbulo superior derecho / si es anterior o posterior)
• Descripción de las características ultrasonográficas del nódulo tiroideo:
Composición del nódulo: Sólido, quístico o espongiforme
Ecogenicidad: Isoecoico, hiperecoico, hipoecoico
Márgenes: Regulares o Irregulares – Definidos como infiltrativos, microlobulados o espiculados
Presencia y tipo de calcificaciones: Marco o microcalcificaciones
Forma: Si el nódulo está más alto que ancho
Vascularidad: Central o periférica
El patrón ultrasonográfico asociado con un nódulo tiroideo confiere un riesgo de malignidad, y combinado con el tamaño del nódulo, guía en la toma de decisiones. El patrón ultrasonográfico de alta sospecha de malignidad incluye nódulos hipoecoicos, sólidos, o nódulos con componentes mixtos (nódulo hipoecoico sólido y parcialmente quístico) con una o más de las siguientes características: márgenes irregulares (infiltrativos, microlobulados), microcalcificaciones, forma más alta que ancha, calcificaciones en el borde del quiste, evidencia de extensión extra tiroidea.
EPIDEMIOLOGY AND ETIOLOGY OF MEDULLARY THYROID CARCINOMA
Medullary thyroid carcinoma (MTC) is a neuroendocrine tumor of the para-follicular cells or C cells of the thyroid gland.
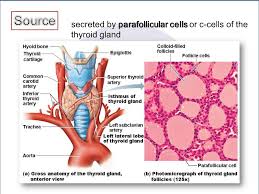
Approximately 1.7% of all thyroid neoplasms are medullary carcinomas. Although most cases are sporadic, 15% to 25% of the cases are part of an autosomal dominant hereditary syndrome.
The production of calcitonin is a characteristic feature of this tumor. The C cells originate in the embryonic neural crest; as a result, medullary carcinomas often have the clinical and histological characteristics of other neuroendocrine tumors such as carcinoids and pancreatic islet cell tumors.
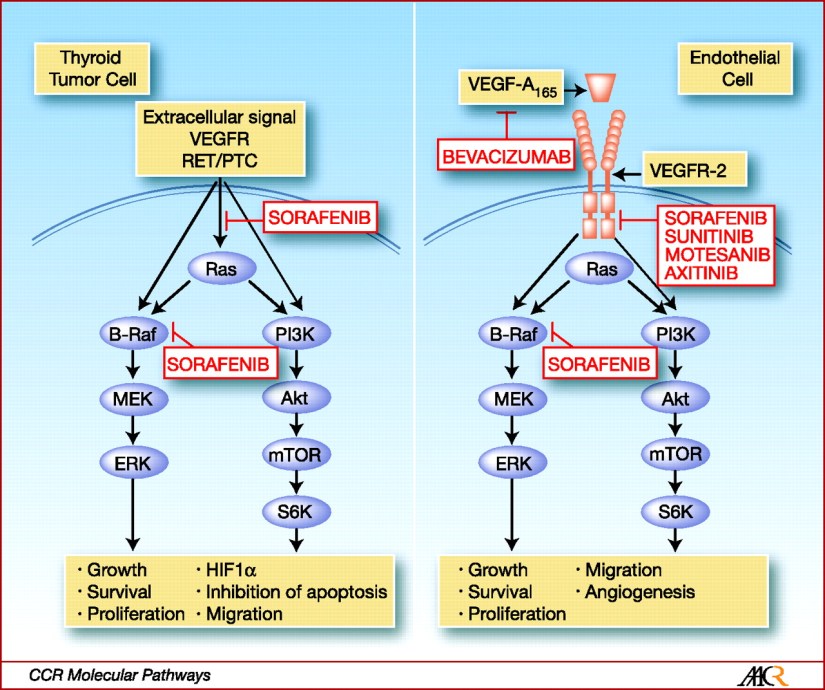
The RET protooncogene (RE- arranged during Transfection) was discovered in 1985 by Takahashi et al. The RET protooncogene is expressed in cells derived from the neural crest, the branchial arches, and the urogenital system. It is located on chromosome 10q11.2, encodes a single-pass transmembrane receptor of the tyrosine kinase family.
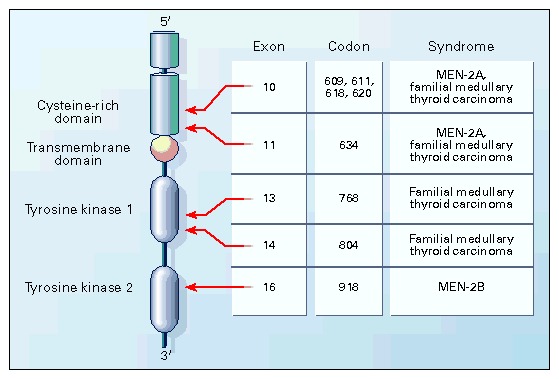
Soon after this discovery it was found that nearly all patients with MEN2A, MEN2B, and familial medullary thyroid cancer (FMTC) have RET germline mutations and roughly 50% of sporadic MTCs have somatic RET mutations.
Researchers recently revealed that 18% to 80% of sporadic MTCs lacking somatic RET mutations have somatic mutations of HRAS, KRAS, or rarely NRAS [17-19].
The probability that an apparently sporadic case of MTC is familial should be considered preoperatively. Despite the absence of a family history of MTC, germline mutations in the RET proto-oncogene have been identified in less than 10% of cases of a seemingly sporadic MTC (index cases) [20, 21].
In sporadic MTC the somatic RET codon M918T mutation appears to indicate an aggressive clinical course and a poor prognosis. Romei C et al reviewed 160 patients with sporadic MTC and found that the prevalence of somatic RET codon M918T mutations varied depending on tumor size: < 1 cm, 11.3% of the cases; 1 to 2 cm, 11.8% of the cases; 2 to 3 cm, 31.8% of the cases; and > 3 cm, 58.8% of the cases. The results of these study raise the question of whether RET acts singlehandedly as the initiator of oncogenesis in sporadic MTC or is activated later as a driver of tumor growth, with other genes playing a significant role in MTC onset. Another reason for these findings is that M918T mutated tumors have a higher growth rate and are more likely to be identified when they are bigger. The low prevalence of the M918T mutation in microcarcinomas may signify a different entity such as carcinoma in situ; precisely because it is not driven by RET.
Several guidelines for the management of sporadic and hereditary MTC have been published by several groups including the North American Neuroendocrine Tumor Society, the National Comprehensive Cancer Network, and the American Thyroid Association (ATA).

These guidelines described the disease phenotypes associated with specific RET mutations in hereditary MTC and recommended timing of early thyroidectomy based on the specific RET mutation. Three of the guidelines used either the TNM designation of the American Joint Committee on Cancer (AJCC), or terms such as Level I, II, or III, or ‘‘high,’’ ‘‘higher,’’ or ‘‘highest,’’ to allocate progressive increases in aggressiveness of the MTC. The criteria used to categorize the aggressiveness of the disease was the development of MTC at an early age, frequently in association with metastatic disease. The original ATA guidelines used A, B, C, and D designations to define categories of RET mutations associated with increasing aggressiveness of the MTC.
Due to significant misunderstanding regarding the different ATA risk categories, the task force recommends that the A and B categories be combined into a new category, ‘‘moderate risk’’ (MOD); category C be changed to a new category, ‘‘high risk’’ (H); and category D be changed to a new category, ‘‘highest risk’’ (HST) [recommendation # 1 from the ATA guidelines). The ATA-HST category includes patients with MEN2B and the RET codon M918T mutation, the ATA-H category includes patients with RET codon C634 mutations and the RET codon A883F mutation, and the ATA- MOD category includes patients with RET codon mutations other than M918T, C634, and A883F. Ever since the discovery of the RET oncogene, over 100 mutations, duplications, insertions, or deletions involving RET protooncogene have been identified in patients with hereditary MTC.
El entrenamiento de Rodrigo Arrangoiz MS, MD, FACS experto en tumores de tiroides fue el siguiente:
• Cirugia general y gastrointestinal:
• Michigan State University:
• 2004 al 2010
• Cirugia oncológica / tumores de cabeza y cuello / cirugia endocrina:
• Fox Chase Cancer Center (Filadelfia):
• 2010 al 2012
• Maestria en ciencias (Clinical research for health professionals):
• Drexel University (Filadelfia):
• 2010 al 2012
• Cirugia de tumores de cabeza y cuello / cirugia endocrina
• IFHNOS / Memorial Sloan Kettering Cancer Center:
• 2014 al 2016
Sin beneficio del Yodo Radioactivo después de la Segunda Cirugía en el Cáncer de Tiroides
Agregar un curso de yodo radiactivo (RAI) a una segunda cirugía para el carcinoma papilar de tiroides (PTC) recurrente o persistente no parece ofrecer un beneficio clínico, y no hay una reducción en la tasa de recurrencias posteriores, concluyen los investigadores estadounidenses.
Sus conclusiones provienen de un análisis retrospectivo de un poco más de 100 pacientes que se sometieron a una reintervención para PTC recurrente.
El estudio mostró que los pacientes experimentaron reducciones en los niveles de marcadores tumorales, ya sea que se sometieran o no a la administración de RAI. No hubo diferencias significativas en las tasas de recurrencia entre los dos grupos.
Sin embargo, el estudio también encontró que hubo menos recurrencias entre los pacientes que se sometieron a cirugía sola en comparación con aquellos que también recibieron terapia RAI, y los receptores de RAI tenían menos probabilidades de tener una excelente respuesta de marcador tumoral.
El estudio fue publicado en línea el 15 de agosto en JAMA Surgery.
El autor del estudio Michael W. Yeh, MD, Sección de Cirugía Endocrina, David Geffen School of Medicine, Universidad de California, Los Angeles (UCLA), dijo que “durante muchos años, se pensó que el yodo radiactivo era solo un regalo de promoción, que se obtiene y casi no hubo efectos secundarios “.
https://www.medscape.com/viewarticle/900880?src=WNL_recnl_180820_MSCPEDIT_hmonc&uac=112600CX&impID=1717418&faf=1#vp_1
El entrenamiento de Rodrigo Arrangoiz MS, MD, FACS experto en tumores de tiroides fue el siguiente:
• Cirugia general y gastrointestinal:
• Michigan State University:
• 2004 al 2010
• Cirugia oncológica / tumores de cabeza y cuello / cirugia endocrina:
• Fox Chase Cancer Center (Filadelfia):
• 2010 al 2012
• Maestria en ciencias (Clinical research for health professionals):
• Drexel University (Filadelfia):
• 2010 al 2012
• Cirugia de tumores de cabeza y cuello / cirugia endocrina
• IFHNOS / Memorial Sloan Kettering Cancer Center:
• 2014 al 2016
No Benefit From RAI After Second Thyroid Cancer Surgery
Adding a course of radioactive iodine (RAI) to a second surgery for recurrent or persistent papillary thyroid carcinoma (PTC) does not appear to offer a clinical benefit, and there is no reduction in the rate of subsequent recurrences, conclude US researchers.
Their conclusions come from a retrospective analysis of just over 100 patients who underwent reoperation for recurrent PTC.
The study showed that patients experienced reductions in levels of tumor markers whether or not they underwent RAI administration. There was no significant difference in recurrence rates between the two groups.
However, the study also found there were fewer recurrences among patients who underwent surgery alone compared to those who also received RAI therapy, and RAI recipients were less likely to have an excellent tumor marker response.
The study was published online August 15 in JAMA Surgery.
Study author Michael W. Yeh, MD, Section of Endocrine Surgery, David Geffen School of Medicine, University of California, Los Angeles (UCLA), said that “for many years, radioactive iodine was thought to be just a freebie, that you got it and there were almost no side effects.”
El entrenamiento de Rodrigo Arrangoiz MS, MD, FACS experto en tumores de tiroides fue el siguiente:
• Cirugia general y gastrointestinal:
• Michigan State University:
• 2004 al 2010
• Cirugia oncológica / tumores de cabeza y cuello / cirugia endocrina:
• Fox Chase Cancer Center (Filadelfia):
• 2010 al 2012
• Maestria en ciencias (Clinical research for health professionals):
• Drexel University (Filadelfia):
• 2010 al 2012
• Cirugia de tumores de cabeza y cuello / cirugia endocrina
• IFHNOS / Memorial Sloan Kettering Cancer Center:
• 2014 al 2016
https://www.medscape.com/viewarticle/900880?src=WNL_recnl_180820_MSCPEDIT_hmonc&uac=112600CX&impID=1717418&faf=1#vp_1
Yodo Radioactivo en el de Cáncer Tiroides
La mayoría (75% o más) de los carcinomas tiroideos de células foliculares (FCDTC) retienen la capacidad de las células foliculares tiroideas normales para absorber y concentrar el yodo.
Esta concentración de yodo es menos eficiente que la observada en la glándula tiroides normales , debido a la arquitectura anormal de las estructuras foliculares dentro del cáncer, dificulta la organificación y retención del isótopo, lo que explica por qué los cánceres se ven típicamente como nódulos “fríos” en las imágenes isotópicas en las gammagrafías tiroideas. Sin embargo, esta función diferenciada retenida permite isótopos radiactivos de yodo para ser utilizados tanto para la localización como para el tratamiento del carcinoma de tiroides residual.
Cuando se administran por vía oral, todos los isótopos de yodo se absorben rápidamente y de manera confiable del tracto gastrointestinal superior, circulan transitoriamente en el torrente sanguíneo y se concentran en tejidos que expresan un transportador funcional de yoduro de sodio (NIS). El isótopo restante se filtra y excreta a través de la vía renal, con la consiguiente exposición a la radiación a todo el tracto urinario. Los tejidos que concentran activamente el yodo incluyen tejido tiroideo normal y canceroso, glándula salival, mama (particularmente durante la lactancia), estómago, riñón y colon.
La absorción de yodo en el tejido tiroideo normal y maligno, aunque no en la mayoría de los otros tejidos, depende de la actividad del receptor de la hormona estimulante de la tiroides (TSH), que regula la expresión y aumenta la activación de NIS en el tejido tiroideo.
Además, el tejido tiroideo es capaz de organificar yodo a tiroglobulina, una reacción que requiere al menos una estructura folicular parcialmente intacta. Dicha organización aumenta la vida media biológica del yodo, aumentando la exposición del tejido tiroideo a la irradiación y mejorando la lesión celular y la muerte celular inducidas por la radiación.
Esos isótopos radiactivos de yodo en uso clínico (131I, 123I) emiten rayos γ, que pueden detectarse utilizando un aparato de detección apropiado (una cámara gamma), permitiendo la formación de imágenes de tejidos que concentran yodo y por lo tanto la detección y localización de residuos o metástasis cáncer de tiroides, después de la estimulación con TSH. Esta técnica de escaneo de todo el cuerpo se convirtió en el pilar de la vigilancia posoperatoria del cáncer de tiroides en América del Norte en los años 80 y 90, aunque se ha utilizado con menos frecuencia en los últimos años, como el mejoramiento de la tecnología de ultrasonido, imágenes transversales y mediciones de la tiroglobulina estimulada ha demostrado ser más sensible y más específica.
Sin embargo, la introducción de la tomografía por emisión de fotón único (SPECT), en particular, para localizar con mayor precisión las áreas de concentración de yodo asegura que las imágenes de isótopos continúen desempeñando un papel útil en la evaluación de pacientes con cáncer de tiroides residual conocido.
Aunque los rayos γ son de alta energía, su absorción en el tejido es baja y la mayoría de estas partículas no interactúan con la célula en la que se concentra el yodo o con el tejido circundante. Aunque esto es óptimo para obtener imágenes, brinda una buena resolución de imagen , los rayos γ no son particularmente efectivos en el tratamiento del carcinoma tiroideo residual, que en cambio depende de la emisión de partículas beta, la principal partícula emitida por la descomposición de 131I pero no de 123I.
Las partículas beta de energía moderadamente alta emitidas por 131I tienen una la longitud mediana y una ruta corta en los tejidos humanos, viajando, en promedio, a solo 0.5 cm antes de interactuar con el tejido circundante.
La ionización resultante causa daño en el ADN, incluyendo roturas de ADN de una y dos cadenas. Esta lesión del ADN es detectada por la célula , activando la ruta de p53, que comúnmente está intacta en las células diferenciadas de carcinoma de tiroides.
Ante un daño menor en el ADN, los mecanismos de reparación celular se activan, por lo general restaurando la célula a la salud normal, aunque con el potencial de la inducción de mutaciones adicionales o reordenamientos cromosómicos.
Sin embargo, con un daño del ADN más extenso, la activación de p53 desencadena la apoptosis (muerte celular programada) de la célula afectada.
Debido a que las células cancerosas -incluido el cáncer de tiroides- a menudo carecen de mecanismos eficientes para reparar las roturas de ADN de doble cadena, hay razón de creer que el cáncer de tiroides residual es susceptible a los efectos de la irradiación beta, más que el tejido normal circundante, aunque todavía no existen datos in vitro, clínicos para apoyar esta hipótesis.
El entrenamiento de Rodrigo Arrangoiz MS, MD, FACS experto en tumores de tiroides fue el siguiente:
• Cirugia general y gastrointestinal:
• Michigan State University:
• 2004 al 2010
• Cirugia oncológica / tumores de cabeza y cuello / cirugia endocrina:
• Fox Chase Cancer Center (Filadelfia):
• 2010 al 2012
• Maestria en ciencias (Clinical research for health professionals):
• Drexel University (Filadelfia):
• 2010 al 2012
• Cirugia de tumores de cabeza y cuello / cirugia endocrina
• IFHNOS / Memorial Sloan Kettering Cancer Center:
• 2014 al 2016
Hiperparatiroidismo Primario Pediatría
El Hiperparatiroidismo Primario (HPTP) es una enfermedad endocrina frecuente en adultos, con una prevalencia del 3/1.000 en la población general, que se eleva hasta 21/1.000 en la mujer postmenopáusica6. En las grandes series, sin embargo, la ocurrencia de HPTP en niños o adolescentes no supera el 2%.
La revisión de la literatura confirma que alrededor de 80% de los HPTP en niños y adolescentes son sintomáticos al momento del diagnóstico.
Los síntomas principales de presentación del HPTP en adolescentes son: fatiga, letargia, cefalea, depresión, nefrolitiasis, dolor abdominal, vómitos, polidipsia, poliuria y retraso en el crecimiento, todos ellos relacionados con hipercalcemia significativa. Muchos de estos síntomas son inespecíficos, lo que probablemente es responsable del diagnóstico tardío. A este respecto cabe destacar que el tiempo medio entre el inicio de los síntomas y el diagnóstico es mayor a dos 2 años como. Por otro lado, en el momento de la presentación aproximadamente 40% de los pacientes jóvenes presenta daño en un órgano blanco (nefrocalcinosis, nefrolitiasis, enfermedad ósea, pancreatitis).
Respecto a la etiología, también hay diferencias en la presentación del HPTP en niños en comparación con los adultos. En niños y adolescentes existe una mayor proporción de formas familiares y por tanto de hiperplasia o enfermedad multiglandular. Así, un estudio muestra que alrededor de 50% corresponden a adenomas, 39% a hiperplasia de las 4 glándulas paratiroides (repartidos entre enfermedad esporádica, neoplasia endocrina múltiple (NEM) tipo I y II A), 3% de carcinoma paratiroideo y en 8% no se encuentra lesión paratiroidea en la primera cirugía. Un hecho destacable y con implicancias terapéuticas es que cuando existe una historia familiar de HPTP ello se relaciona fuertemente con hiperplasia de paratiroides y excepcionalmente con adenomas.
El diagnóstico bioquímico del HPTP en el adolescente no presenta diferencias respecto del que se realiza en el adulto, basándose en la presencia de hipercalcemia, en concomitancia con PTH elevada (85% de las veces) o inapropiadamente normal (15% restante), asociada a hipercalciuria. A diferencia del adulto, la determinación rutinaria de calcemia es menos frecuente y por ello requiere de un mayor índice de sospecha, recomendándose su determinación cuando se está frente a uno de lo síntomas o complicaciones mencionadas.
En cuanto a la utilidad de los métodos de imagen en el HPTP hay controversia respecto a su uso rutinario en el preoperatorio de pacientes adultos, dado que existe una tasa no despreciable de falsos positivos y negativos y se ha establecido el concepto que el “mejor método de localización” de la lesión paratiroidea en pacientes operados por primera vez, es un cirujano experto.
No obstante, en la edad pediátrica, se sugiere realizar de rutina estudios de imagen complementarios en el preoperatorio debido a la alta tasa de hiperplasias paratiroideas y adenomas ectópicos, lo que se asocia a una elevada frecuencia de reoperaciones (20%-25%).
Un estudio mostró 9% de adenomas paratiroideos mediastínicos en edad pediátrica y en estos casos la cintigrafía sería de especial utilidad dado que identificaría 100% de estas localizaciones extracervicales. La ultrasonido cervical presenta una sensibilidad de 86%, especificidad de 67% y valor predictivo positivo de 95%. De esta forma, los estudios imagenológicos recomendados son el cintigrama MIBI-SPECT y la ultrasonido cervical de alta resolución.
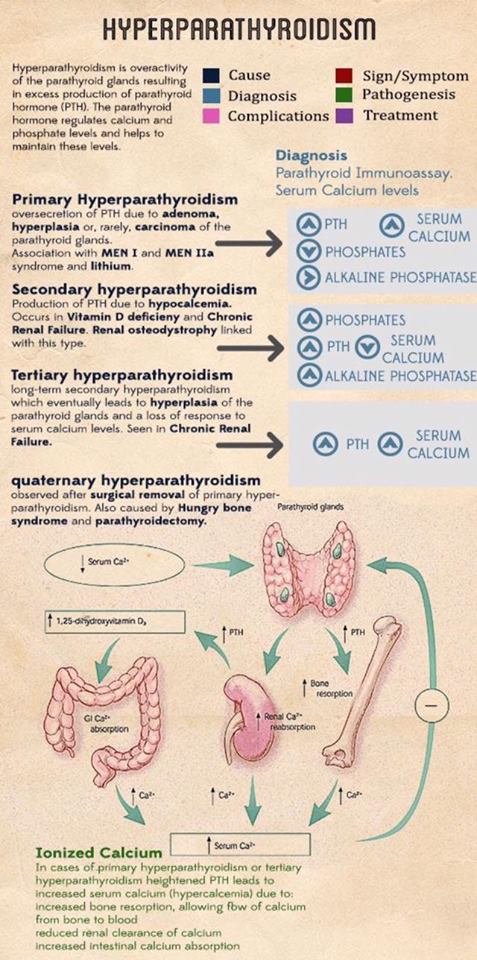
Primary Hyperparathyroidism
The overproduction of parathyroid hormone (PTH), termed hyperparathyroidism (HPT), can be categorized as primary, secondary, or tertiary. Primary hyperparathyroidism (PHPT) arises from an unregulated overproduction of PTH from an abnormal parathyroid gland. Increased PTH levels may also occur as a compensatory response to hypocalcemic states resulting from chronic renal failure or gastrointestinal (GI) malabsorption of calcium. This secondary HPT can be reversed by correction of the underlying problem (e.g., kidney transplantation for chronic renal failure). However, chronically stimulated parathyroid glands may occasionally become autonomous, resulting in persistence or recurrence of the hypercalcemia after successful renal transplantation, resulting in tertiary HPT.
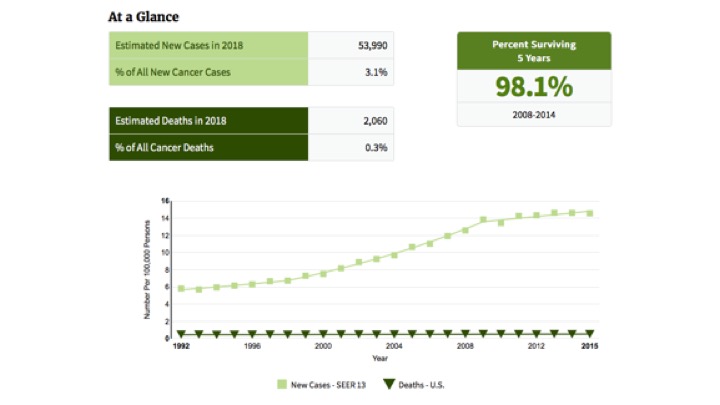
Epidemiology and Etiology
PHPT is defined as hypercalcemia or widely fluctuating levels of serum calcium resulting from the inappropriate or autogenous secretion of PTH by one or more parathyroid glands in the absence of a known or recognized stimulus.
The most common cause of hypercalcemia in the outpatient setting is PHPT, with approximately 100,000 new cases per year reported in the United States.
Since the advent of routine laboratory testing, the prevalence of the disease has increased from 0.1% to 0.4% (one to seven cases per 1000 adults).
In a study by Yeh et al, the incidence of PHPT fluctuated between 36.3 and 120.2 cases per 100,000 women-year and 13.4 and 35.6 in 100,000 men-year. PHPT may present at any age, with the vast majority of cases occurring in patients older than 45 years of age. The mean age at diagnosis has remained between 52 and 56 years.
Women have consistently made up the preponderance of cases, with a female-to-male ratio of 3 to 4 : 1. Based on a population based study from Rochester Minnesota the higher incidence of this could be secondary (hypothetically) to estrogen deficiency after menopause that reveals underlying HPT.
The precise origin of PHPT is unknown, although exposure to low-dose therapeutic ionizing radiation and familial predisposition account for some cases.
Irradiation for acne could have accounted for a 2 to 3-fold increase in the incidence of this disease at some point in time, and a 4-fold increase was noted in survivors of the atomic bomb.
Schneider et al, in their study of 2555 patients followed for 50 years, even low doses of radiation exposure during the teenage years was associated with a slight risk of developing PHPT . In this study a dose response was documented in people receiving external-beam radiotherapy for benign diseases before their 16th birthday.
The latency period for the development of PHPT after radiation exposure is longer than that for the development of thyroid tumors, with most cases occurring 30 to 40 years after exposure.
Patients who have been radiated have similar clinical manifestations and serum calcium levels when compared to patients without a history of radiation exposure. However, the former tend to have higher PTH levels and a higher incidence of concomitant thyroid neoplasms .
Certain medications have been implicated in the development of hypercalcemia. Lithium therapy has been known to shift the set point for PTH secretion in parathyroid cells, thereby resulting in elevated PTH levels and mild hypercalcemia.
Lithium stimulates the growth of abnormal parathyroid glands in vitro and also in susceptible patients in vivo 16. Unusual metabolic features associated with lithium use include low urinary calcium excretion, normal cyclic AMP excretion and lack of calcic nephrolithiasis. The mechanism probably results from lithium linking with the calcium sensing receptor on the parathyroid glands resulting in PTH secretion.
Elevated serum calcium levels have been associated with thiazide diuretic. The overall annual age- and sex-adjusted (to 2000 U.S. whites) incidence was 7.7 (95% CI, 5.9 to 9.5) per 100,000 individuals.
The average 24-hour plasma calcium concentrations are increased with thiazide diuretic use, but the mean 24-hour PTH levels remain unchanged in subjects with normal baseline PTH levels and no evidence of hypercalciuria.
Thiazides diuretics have several metabolic effects that may contribute to increased calcium levels. A decrease in urine calcium excretion is the most likely cause, but in some cases diuretic use has been associates with a metabolic alkalosis that could also cause an increase in total serum calcium levels through a pH-dependent increase in protein-bound calcium.
Although plasma 1,25 (OH) vitamin D levels are unchanged, increased intestinal calcium absorption in response to thiazide diurectic use has been noted and could also contribute to an increase in serum calcium. One last possible explanation for the elevated serum calcium levels associated with thiazide diuretic use is hemoconcentration associated with diuresis.
Numerous genetic abnormalities have been identified in the development of PHPT, including anomalies in tumor suppressor genes and proto-oncogenes. Specific DNA mutations in a parathyroid cell may confer a proliferative advantage over normal neighboring cells, thus allowing for clonal growth. Large populations of these altered cells containing the same mutation within hyper functioning parathyroid tissue suggest that such glands are a result of clonal expansion.
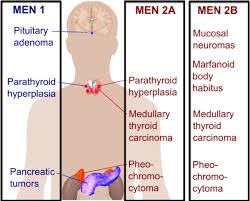
The majority of PHPT cases are sporadic. Nonetheless, PHPT also occurs within the spectrum of a number of inherited disorders such as multiple endocrine neoplasia syndromes (MEN), MEN type 1 (Wermer Syndrome), MEN type 2A (Sipple Syndrome), isolated familial HPT, and familial HPT with jaw-tumor syndrome. All of these syndromes are inherited in an autosomal dominant fashion.
The earliest and most common presentation of MEN 1 is PHPT, and develops in approximately 80% to 100% of patients by age 40 years. These patients also are predisposed to the development of pancreatic neuroendocrine tumors and pituitary adenomas and, less frequently, to skin angiomas, lipomas, adrenocortical tumors, and neuroendocrine tumors of the thymus, bronchus, or stomach. MEN type 1 has been shown to result from a germline mutation in a tumor suppressor gene, called MEN1 gene, located on chromosome 11q12-13 that encodes Menin, a protein that is postulated to interact with the transcription factors JunD and nuclear factor-κB in the nucleus, in addition to replication protein A and other proteins.
Pre-symptomatic screening for mutation carriers for MEN type 1 is difficult because generally MEN1 mutations result in a nonfunctional protein and are scattered throughout the translated nine exons of the gene. MEN1 mutations also have been found in kindred’s initially suspected to represent isolated familial HPT. Screening for mutation carriers for MEN type 1 has a very high detection rate, greater than 94%, and is used in Sweden for patients with PHPT with a first-degree relative with a major endocrine tumor, age of onset is less than 30 years and/or if multiple pancreatic tumors / parathyroid hyperplasia is detected; thus these patients should be screened for MEN1 mutations.
Approximately 20% of patients with MEN type 2A (Sipple Syndrome) develop PHPT which is usually less severe. MEN type 2A is caused by a germline mutation of the RET proto-oncogene located on chromosome 10. Genotype and phenotype correlations have been noted in this syndrome in that individuals with mutations at codon 634 are more likely to develop PHPT. Patients with the familial HPT with jaw-tumor syndrome have an increased predisposition to parathyroid carcinoma. This syndrome maps to a tumor suppressor locus HRPT2 (parafibromin) on chromosome 1.
Sporadic parathyroid adenomas and some hyperplastic parathyroid glands have loss of heterozygosity (LOH) at 11q13, the site of the MEN1 gene in approximately 25% to 40% of the cases. Overexpression of PRAD1, which encodes cyclin D1, a cell cycle control protein, is found approximately 18% of parathyroid adenomas 40, 41. This was proven to result from a rearrangement on chromosome 11 that places the PRAD1 gene under the control of the PTH promoter.
Other chromosomal regions deleted in parathyroid adenomas and possibly reflecting loss of tumor suppressor genes include 1p, 6q, and 15q, whereas amplified regions suggesting oncogenes have been identified at 16p and 19p .
RET mutations are unusual in sporadic parathyroid tumor. Sporadic parathyroid cancers are characterized by uniform loss of the tumor suppressor gene RB, which is involved in cell cycle regulation, and 60% have HRPT2 (CDC73) mutations. These alterations are rare in benign parathyroid tumors and may have implications for diagnosis. The p53 tumor suppressor gene is also inactivated in a subset (30%) of parathyroid carcinomas.
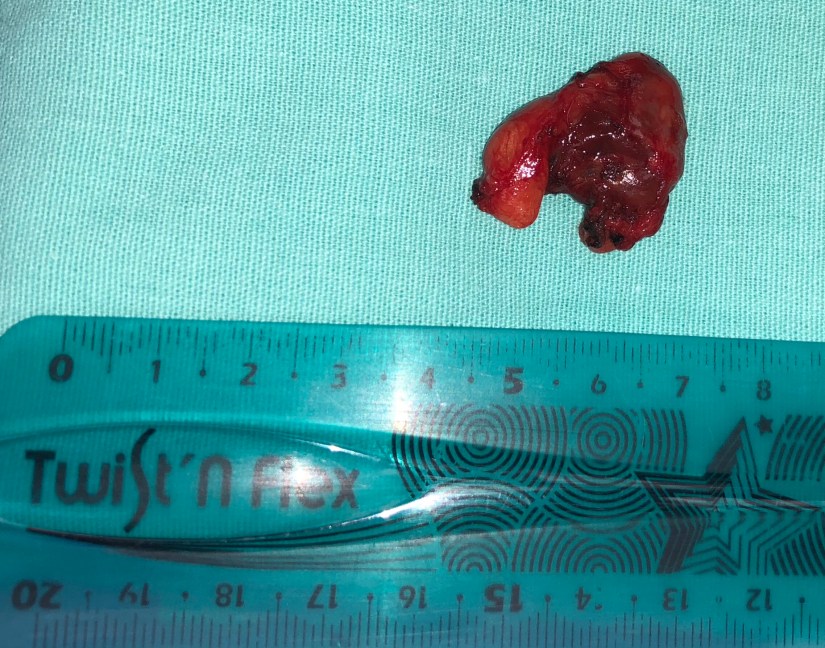
Single gland adenoma is the most common cause (75% to 85%), lower pole adenomas (in relation to the thyroid) are more common than are upper pole adenomas; sizes range from 1 cm to 3 cm.

The normal weight of a parathyroid gland is approximately 40 to 50 mg, and the weight of parathyroid adenomas vary between 553.7 +/- 520.5 mg (range, 66-2536). Ectopic glands can be present (4% to 16% of cases).

PHPT is caused by the enlargement of a single parathyroid gland or parathyroid adenoma in approximately 75% to 89% of the cases, multiple adenomas or hyperplasia in 15% to 25% of the cases, and parathyroid carcinoma as the cause of PHPT is extremely rare in most parts of the world (~1%) of patients. Multi-gland adenoma arises in a significant number of patients, double adenomas are seen in approximately 2% to 12% of the cases, triple adenomas in less than 1% the cases, and four adenomas or parathyroid gland hyperplasia in less than 3% to 15% of the cases.

Most parathyroid adenomas consist of parathyroid chief cells. They are usually encapsulated and in 50% of the cases they are surrounded by normal parathyroid tissue. Some adenomas, nevertheless, are composed of oxyphil cells. These adenomas are usually larger than chief cell adenomas.
Parathyroid adenomas are sometimes located within the thymus and they express a parathyroid-specific gene, GCMB, contrasting with the normal thymus, which does not neither PTH nor GCMB.
In a study by Ruda et al, 20,225 patients with PHPT, parathyroid hyperplasia accounted for approximately six percent of cases. In parathyroid hyperplasia all four glands are enlarged, with the lower glands typically being larger than the upper ones. The glands are usually composed of chief cells. Clear cell hyperplasia is very rare, and is the only form in which the upper glands are larger than the lower ones.
The training of Rodrigo Arrangoiz MS, MD, FACS expert in thyroid tumors was as follows:
• General and gastrointestinal surgery:
• Michigan State University:
• 2004 to 2010

• Surgical oncology / head and neck tumors / endocrine surgery:
• Fox Chase Cancer Center (Philadelphia):
• 2010 to 2012

• Master of Science (Clinical research for health professionals):
• Drexel University (Philadelphia):
• 2010 to 2012

• Head and Neck Surgeon / Endocrine Surgery
• IFHNOS / Memorial Sloan Kettering Cancer Center:
• 2014 to 2016

TRATAMIENTO DEL CÁNCER DE TIROIDES

El tratamiento de los tumores tiroideos, y en algunos casos es la resección quirúrgica. El objetivo del manejo del cáncer tiroideo sigue siendo la eliminación por completo de la enfermedad con una mínima morbilidad.
-
El tratamiento quirúrgico adecuado permitirá un seguimiento postoperatorio cuidadoso, terapias adyuvantes si es necesario, y minimiza la posibilidad de recurrencia de la enfermedad.
La cirugía para el cáncer de tiroides es un elemento vital de un enfoque de tratamiento multifacético. La operación recomendada debe ser compatible con la estrategia general de manejo y el plan de seguimiento recomendados por el equipo multidisciplinario.
-
Se debe considerar derivar a los pacientes con características de alto riesgo (enfermedad clínica N1, preocupación por la invasión del nervio recurrente laríngeo, o enfermedad extremadamente invasiva) a cirujanos experimentados, ya que tanto la calidad de la cirugía como la experiencia del cirujano puede tener un impacto significativo en los resultados clínicos y las tasas de complicaciones.
Debido a que el cáncer papilar de tiroides tiene una tasa de mortalidad extremadamente baja, la recurrencia de la enfermedad se ha convertido en el principal objetivo de interés a la hora de decidir el manejo quirúrgico óptimo para la mayoría de los pacientes.
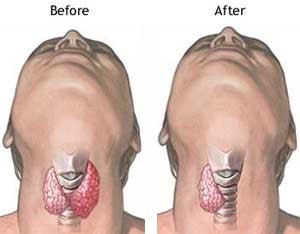
Para los pacientes con cáncer papilar de tiroides que miden más de 1 cm, la cirugía que históricamente se ha recomendado es una tiroidectomía total que ciertamente sigue siendo la operación apropiada para los cánceres de tiroides bien diferenciados de alto riesgo.
-
Las razones que se utilizan para considerar la realización de una tiroidectomía total en los carcinoma tiroideos de bajo riesgo incluyen lesiones identificadas dentro del lóbulo tiroideo contralateral debido a que focos de cáncer papilar de tiroides se encuentran de manera bilateral en hasta 85% de los casos y en un 5% a 10% de los casos de recurrencia el foco de recurrencia se encuentra en el lóbulo contralateral cuando se realiza una lobectomía tiroidea.
-
Desde el punto de vista postoperatorio, el tejido tiroideo que queda, si se realiza una resección más conservadora, hace prohibitiva la ablación con yodo radiactivo de la glándula restante. Además, la medición sérica de la tiroglobulina como un marcador de enfermedad persistente o recurrente después de la lobectomía tiroidea es más difícil de interpretar dado el tejido tiroideo restante. Una tiroidectomía total evita estas dificultades y minimiza la cirugía re operativa que se asocia con un aumento de las tasas de complicaciones.
Si se elige tratamiento quirúrgico para los pacientes con cáncer de tiroides menores a un 1 cm sin extensión extra tiroidea y sin evidencia clínica de metástasis ganglionares (cN0), el procedimiento quirúrgico inicial debe ser una lobectomía tiroidea a menos que haya indicaciones claras para extirpar el lóbulo contralateral (recomendación 35 del ATA).
-
La lobectomía tiroidea es un tratamiento adecuado para los carcinomas intratiroideos pequeños, unifocales, en ausencia de radiación previa a la cabeza y cuello, carcinomas tiroideos familiares, o metástasis ganglionares cervicales clínicamente detectables (recomendación 35 del ATA). Siempre se debe de tomar en cuenta la preferencia del paciente durante la discusión del tratamiento.
Para pacientes con cáncer de tiroides mayores a 1 cm y menores 4 cm sin extensión extra tiroidea, y sin evidencia clínica de metástasis ganglionares (cN0), el procedimiento quirúrgico inicial puede ser un procedimiento bilateral (tiroidectomía casi total o total) o un procedimiento unilateral (lobectomía) (recomendación 35 del ATA).
-
La lobectomía tiroidea puede ser el tratamiento inicial para los carcinomas papilares y foliculares de bajo riesgo; sin embargo, el equipo manejando al paciente puede elegir la tiroidectomía total para permitir el tratamiento con yodo radioactivo o para facilitar el seguimiento de estos pacientes (recomendación 35 del ATA). Siempre se debe de tomar en cuenta la preferencia del paciente durante la discusión del tratamiento.
Existe controversia sobre si se debe realizar y la extensión de disección profiláctica de los ganglios linfáticos con el fin de prevenir la recurrencia local, proporcionar una estadificación más precisa, y aumentar la supervivencia. La distinción entre una disección del compartimento central terapéutico versus profiláctico (o electivo) es que una disección terapéutica implica que la enfermedad ganglionar ya se ha producido y se ha detectado clínicamente o mediante imágenes preoperatorias (enfermedad cN1).

-
Una disección del compartimento central electiva o profiláctica implica que no hay evidencia clínica o radiográfica de metástasis ganglionares. Esta diferencia es importante porque el impacto de tener ganglios linfáticos clínicamente detectables en la supervivencia y la recidiva local puede diferir comparado con enfermedad detectada de manera microscópica. Igualmente, una disección del compartimiento central puede ser ipsilateral (el mismo lado que el tumor dominante) o bilateral (ipsilateral y contralateral) y es importante documentar esta distinción en la nota quirúrgica.
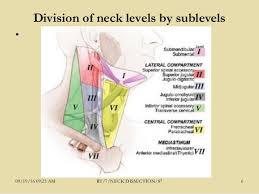
El compartimento central (nivel VI) esta limitado superiormente por el hueso hioides, inferiormente por la arteria innominada, y lateralmente por las arterias carótidas. La disección terapéutica del compartimento central (nivel VI del cuello) para los pacientes con ganglios centrales clínicamente involucrados debe acompañar a la tiroidectomía total para proporcionar la resección completa de la enfermedad (recomendación 36 del ATA).
Se debe considerar la disección preventiva / profiláctica del compartimiento central (ipsilateral o bilateral) en pacientes con carcinoma papilar de tiroides con ganglios linfáticos clínicamente no involucrados (cN0) en pacientes con tumores primarios avanzados (T3 o T4), o ganglios linfáticos clínicamente comprometidos en el compartimiento lateral del cuello (cN1b), o si la información se usará para planificar pasos adicionales en la terapia (recomendación 36 del ATA).
La tiroidectomía sin disección profiláctica del compartimiento central es apropiada para tumores papilares pequeños (T1 o T2), no invasivas, con ganglios linfáticos clínicamente negativos (cN0) y para la mayoría de los cánceres foliculares (recomendación 36 del ATA).
La disección terapéutica de los ganglios linfáticos del compartimiento lateral debe realizarse en pacientes con linfadenopatía cervical lateral metastásica comprobada por biopsia (recomendación 37 del ATA). La extirpación aislada de los ganglios linfáticos afectados, conocida como “berry picking,”, viola el compartimento central sin abordar adecuadamente toda la extensión de la enfermedad y puede estar asociado con mayores tasas de recurrencia y morbilidad en la cirugía de revisión.
Por lo general, el diagnóstico de un carcinoma de células foliculares o de Hürthle se realiza después del procedimiento quirúrgico que generalmente es una lobectomía tiroidea. En estas circunstancias, a menudo se realiza una tiroidectomía total en pacientes de alto riesgo cuando se anticipa que el paciente va a requerir tratamiento adyuvante con yodo radioactivo, ya que todo el tejido tiroideo debe ser eliminado para que el yodo radioactivo sea efectivo.
Se debe ofrecer le a los pacientes que se le realizó una lobectomía tiroidea completar la tiroidectomía total a los pacientes que se habría recomendado una tiroidectomía bilateral si el diagnóstico hubiera estado disponible antes de la cirugía inicial (recomendación 38 del ATA). La disección terapéutica de los ganglios linfáticos del compartimiento central debe incluirse si los ganglios linfáticos están clínicamente involucrados (recomendación 38 del ATA).
La lobectomía tiroidea por si sola puede ser considerada como un manejo suficiente para los carcinomas papilares y foliculares de bajo riesgo (recomendación 38 del ATA). La ablación con yodo radiactivo en lugar de completar la tiroidectomía no se recomienda de forma rutinaria; sin embargo, se puede usar para quemar el lóbulo remanente en casos seleccionados (recomendación 38 del ATA).
El carcinoma anaplásico representa un desafío único porque rara vez se diagnostica de manera oportuna, por lo que el manejo quirúrgico generalmente solo se ofrece como una opción paliativa.
-
En el caso raro en que el carcinoma anaplásico haya sido diagnosticado incidentalmente o al inicio de su evolución, la tiroidectomía total con linfadenectomía del compartimiento central y linfadenectomía radical modificada ipsilateral ofrece la mejor oportunidad de supervivencia en el caso excepcional de que el tumor sea intratiroideo.
-
Dada la naturaleza agresiva y la supervivencia limitada para los pacientes con carcinoma anaplásico, a menudo se evita la intervención quirúrgica agresiva que implica la resección de estructuras adyacentes, como la laringe, la faringe o el esófago, debido a la morbilidad excesiva asociada.
-
La resección de enfermedad que se extiende más allá de la glándula tiroides puede ser apropiada en individuos altamente seleccionados como parte de un régimen de tratamiento multimodal junto con radiación, quimioterapia, y inmunoterapia.


El entrenamiento de Rodrigo Arrangoiz MS, MD, FACS experto en tumores de tiroides fue el siguiente:
• Cirugia general y gastrointestinal:
• Michigan State University:
• 2004 al 2010
• Cirugia oncológica / tumores de cabeza y cuello / cirugia endocrina:
• Fox Chase Cancer Center (Filadelfia):
• 2010 al 2012
• Maestria en ciencias (Clinical research for health professionals):
• Drexel University (Filadelfia):
• 2010 al 2012
• Cirugia de tumores de cabeza y cuello / cirugia endocrina
• IFHNOS / Memorial Sloan Kettering Cancer Center:
• 2014 al 2016
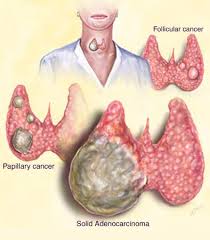
Cáncer de Tiroides
Los nódulos tiroideos son un problema importante de salud pública. Los estudios epidemiológicos han demostrado que la prevalencia de los nódulos tiroideos palpables es aproximadamente del 5% en mujeres y del 1% en hombres que viven en partes del mundo con suficiente yodo.