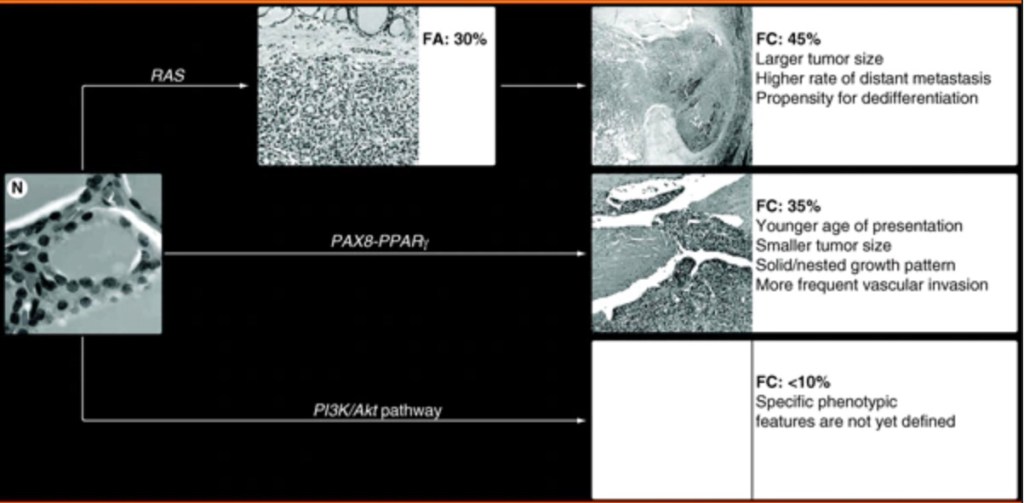
The most frequent genetic alterations in follicular thyroid carcinomas (FTC) include point mutations of the RAS genes and PAX8 / PPARγ rearrangement (Figure).
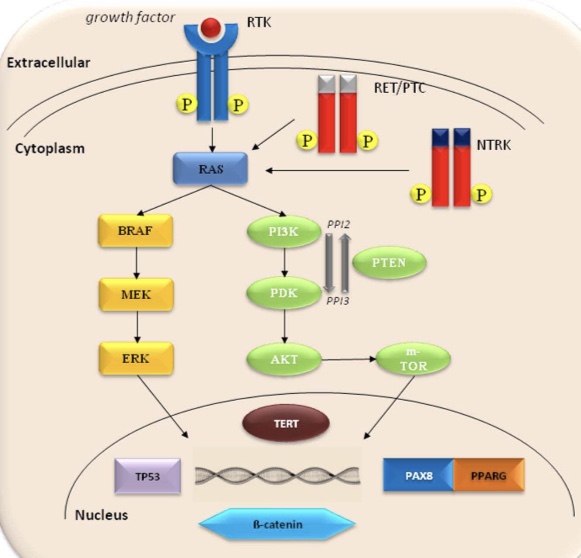
In addition, mutations and other alterations of the genes coding for the effectors of the PI3K/AKT signaling pathway can be found in these tumors, although the frequency of those mutations is low and their biological and diagnostic value remains to be elucidated.
As RAS point mutations are also frequently found in follicular adenomas (FA), it is likely that follicular thyroid carcinomas may arise as a result of malignant transformation of follicular adenomas
RAS mutations are found in 40% to 50% of conventional follicular thyroid carcinomas and in 20% to 40% of adenomas.
In adenomas, the mutations are more common in tumors with a microfollicular growth pattern. Most frequently affected hotspots are NRAS codon 61 and HRAS codon 61.
A lower incidence of RAS mutation has been reported in oncocytic (Hurthle cell) tumors, where only 0% to 4% of adenomas and 15% to 25% of carcinomas appeared to be affected.
The presence of RAS mutation in follicular thyroid carcinomas has been found to correlate with tumor dedifferentiation and a less favorable prognosis.
Several studies have found a significant correlation between RAS mutation and metastatic behavior of follicular thyroid carcinomas, especially with respect to bone metastases.
The more aggressive biological properties of these tumors may be due to the effect of the mutant RAS protein on promoting chromosomal instability, which has been demonstrated al least in the in vitro setting.
The increasing chromosomal instability may predispose the tumor cells for acquiring additional mutations which would result in more malignant phenotype.
The diagnostic use of RAS mutation detection is controversial. On the one hand, it is not specific for malignancy since RAS mutations also occur with significant prevalence in benign follicular adenomas. On the other hand, RAS mutations frequently occur in follicular thyroid carcinomas and the follicular variant papillary carcinomas, both of which are difficult to diagnose cytologically in thyroid FNA samples. Moreover, since mutant RAS is likely to predispose to progression from follicular adenoma to follicular thyroid carcinoma and to further tumor dedifferentiation, it may be justifiable to surgically remove the RAS-positive adenomas to prevent such a progression.
In a prospective study aimed to assess the role of detection of different mutations in improving the preoperative FNA diagnosis of thyroid nodules, the detection of RAS mutations was found to improve the diagnostic accuracy and allowed to diagnose malignant tumors in several samples with negative or insufficient cytology.
PAX8 / PPARγ rearrangement is a result of the translocation t(2;3)(q13;p25). It leads to the fusion between the PAX8 gene, which encodes a thyroid-specific paired domain transcription factor, and the PPARγ gene.
PAX8 / PPARγ occurs in approximately 35% of conventional follicular thyroid carcinomas, and with lower prevalence in oncocytic (Hurthle cell) carcinomas.
Tumors harboring PAX8 / PPARγ tend to present at a younger age, be smaller in size, have a solid / nested growth pattern and more frequently reveal vascular invasion.
The rearrangement results in overexpression of the PPARγ protein that can be detected by immunohistochemistry. However, only strong diffuse nuclear staining correlates with the presence of rearrangement.
PAX8 / PPARγ can also be found in a small fraction (2% to 10%) of follicular adenomas. It has been suggested that follicular adenomas positive for this rearrangement may in fact be pre-invasive (in situ) follicular carcinomas or tumors where invasion was overlooked during histological examination.
The mechanisms of cell transformation induced by PAX8 / PPARγ are not fully understood. Some evidence has been presented for inhibition of normal PPARγ function via a dominant negative effect of the PAX8 / PPARγ protein on wild-type PPARγ. Other studies have found the activation of known PPAR target genes in tumors harboring PAX8 / PPARγ, arguing against the dominant negative effect.
Other possible mechanisms include deregulation of PAX8 function, known to be critical for thyroid cell differentiation, and activation of a set of genes related to neither wild-type PPARγ nor wild-type PAX8 pathways. The lack of understanding of the definitive mechanism of PAX8 / PPARγ action hampers its use as a therapeutic target for follicular thyroid carcinomas, despite the fact that modulators of the PPAR pathway (retinoic acid derivatives as well as thiazolidenediones) are available and have already been used for other diseases.
The detection of PAX8 / PPARγ rearrangement may be of diagnostic value since it occurs almost exclusively in follicular thyroid carcinomas.
It can be achieved by reverse transcriptase-PCR, FISH, or immunohistochemistry with PPARγ antibody. Immunohistochemical detection might be challenging to set up and validate since not all commercially available PPARγ antibodies give a reliable result. The identification of PAX8 / PPARγ rearrangement by reverse-transcriptase PCR or FISH, or finding strong diffuse PPARγ inmmunoreactivity in tumor cells during pathological evaluation should justify the submission of additional sections of the tumor capsule and obtaining deeper levels of all suspicious areas in search for capsular or vascular invasion.
The PI3K / Akt signaling pathway plays an important role in the regulation of cell growth, proliferation and survival. This pathway can be activated by the upstream stimulatory molecules (i.e., RAS, RET /PTC), through the loss of function of PTEN protein that normally inhibits PI3K signaling or as a result of activating mutations or amplification of genes coding for the effectors of this pathway.
The PIK3CA gene, coding for a catalytic subunit of PI3Ks, has been shown to harbor mutations in thyroid tumors, although at low frequency. Specifically, it has been found in 6% to 13% of follicular thyroid carcinomas and in 0% to 6% of follicular adenomas. Mutations typically involve various nucleotides in exons 20 and 9 of the PIK3CA gene.
Mutations of the PTEN gene have been reported in a small proportion of follicular thyroid carcinomas (~7%), but not in follicular adenomas.
Loss of heterozygosity (LOH) of chromosomal regions harboring different tumor suppressor genes is another genetic alteration found in follicular carcinomas and adenomas. The most frequently deleted chromosomal regions in these tumors are on chromosomes 2p, 3p, 9q, 9p, 10q, 11p, 15q and 17p. The average rate of allelic loss of the target regions is significantly higher in follicular carcinomas (30% to 50%) than in follicular adenomas (6% to 15%).
Some studies have found correlation between the frequency of LOH and tumor aggressiveness and outcome in patients with follicular carcinomas. The minimally invasive tumors had overall lower frequency of allelic loss (~30%) compared with the widely invasive follicular carcinomas (~50%). In addition, higher frequency of LOH was associated with disease recurrence.
Allelic loss of the VHLgene on 3p26 was highly specific for malignancy and associated with death from disease in one study of a small series of follicular carcinomas, suggesting that it may serve as an important diagnostic and prognostic marker for follicular carcinomas. However, its clinical utility has to be validated in a larger series of tumors.
Oncocytic (Hurthle cell) neoplasms usually reveal a comparable or even higher rate of LOH than conventional follicular tumors. In oncocytic carcinomas, the most frequently lost regions were on chromosomes 3q and 18q in one study, and on chromosomes 1q, 2p, 8q and 14q in another observation. In the latter report, chromosomal loci at 1q and 2p showed a significantly higher rate of LOH in oncocytic carcinomas than in adenomas, with a 100% sensitivity and 65% specificity in the detection of malignant tumors.
Somatic point mutations and large deletions in mitochondrial DNA have been found in a significant proportion of oncocytic neoplasms. These mutations occur in oncocytic adenomas and carcinomas, and with lower frequency in other types of thyroid neoplasms, including papillary thyroid carcinoma and conventional follicular thyroid carcinoma. More recently, somatic mutations of the GRIM-19 gene have been identified in sporadic oncocytic tumors. GRIM-19 encodes a protein linked to retinoid-interferon-induced pathway of cell death and also involved in mitochondrial metabolism. Mutations in GRIM-19 were found in 15% of oncocytic carcinomas, but were not detected in other types of thyroid cancer, suggesting that alteration of GRIM-19 may serve as a specific marker of oncocytic tumors.
