Upcoming 2025 ATA Guidelines may further support TL in more intermediate-risk patients, especially with molecular profiling and personalized risk stratification
Role of genomics and molecular markers (e.g., BRAF, TERT, RAS) in guiding extent of surgery is under investigation
Conclusions:
Thyroid lobectomy is oncologically safe in selected patients with low-risk DTC (unifocal, ≤ 4 cm, cN0, no ETE or aggressive histology)
Total thyroidectomy remains necessary for high-risk features, RAI candidates, or bilateral disease
The trend is toward individualized, risk-adapted surgical strategies balancing recurrence risk with surgical morbidity
References:
Mazzaferri EL, Young RL. Am J Med. 1981;70(3):511–518.
Bilimoria KY, et al. Ann Surg. 2007;246(3):471–479.
Adam MA, et al. J Clin Oncol. 2014;32(23):2000–2005.
Nixon IJ, et al. Ann Surg. 2012;256(3):518–520.
Sugitani I, et al. World J Surg. 2010;34(6):1215–1221.
Jeon MJ, et al. J Clin Endocrinol Metab. 2017;102(6):1965–1972.
Sanabria A, et al. Cochrane Database Syst Rev. 2020;12:CD012703.
Designed to block aberrant RET signaling in cancers with RET mutations or fusions
It’s FDA-approved for:
Adult and pediatric patients (≥ 2 years):
With advanced / metastatic RET fusion–positive thyroid cancer that is radioactive iodine (RAI)-refractory and requires systemic therapy
Also approved for RET-mutant medullary thyroid cancer and RET fusion–positive non–small cell lung cancer
Clinical Evidence:
LIBRETTO‑001 Trial (RET Fusion –Positive Thyroid Cancer Cohorts):
Participants:
65 adult patients with RET fusion + DTC, including both RAI – refractory and treatment – naïve individuals
Efficacy:
Previously treated cohort (n=41): ORR 85% (95% CI: 71–94%), median duration of response (DOR) 26.7 months
Treatment-naïve cohort (n=24): ORR 96%, median DOR not yet reached (≥ 42.8 months)
Progression-free survival for RET fusion – positive thyroid cancers:
Estimated around 22 months, consistent with real-world data
Safety Profile:
Common side effects include:
Hypertension
Dry mouth
Diarrhea
Fatigue
Edema
Rash
Laboratory abnormalities like elevated ALT / AST
Most AEs were grade 1 to 2, with manageable toxicity:
Low discontinuation rates (~ 2%)
Emerging Roles:
Neoadjuvant Use and Radioactive Iodine Re-sensitization:
Active clinical trials (e.g., NCT04759911, NCT06458036) are evaluating selpercatinib as neoadjuvant therapy prior to surgery or RAI in RET fusion–positive DTC, with the goal to shrink tumors and enhance iodine uptake
Clinical Implications:
First-line option for advanced / metastatic RET fusion + DTC, especially when RAI has failed or is inapplicable
Offers exceptional response rates and durable remissions
Potential for earlier use, including neoadjuvant settings or combination with RAI, pending trial results
Recommended tumor genomic profiling for all advanced thyroid cancers to identify RET alterations and enable targeted therapy
Future Outlook:
Neoadjuvant and combinatorial strategies may broaden selpercatinib’s role in DTC
Ongoing trials will clarify its utility in enhancing RAI responsiveness and enabling less extensive surgery
New category for tumors with high mitotic rate or necrosis:
But that retain differentiation (e.g., follicular or papillary histology)
NIFTP is formally recognized as a low-risk neoplasm, not carcinoma
References:
WHO Classification of Endocrine and Neuroendocrine Tumours (5th Edition, Volume 10), IARC, 2022. https://publications.iarc.fr/
Baloch ZW, LiVolsi VA, Asa SL, et al. Overview of the 2022 WHO Classification of Thyroid Tumors. Endocr Pathol. 2022;33(1):27–63. https://doi.org/10.1007/s12022-022-09712-6
Lam AK. Update on WHO classification of thyroid tumors in 2022. Histopathology. 2022;81(4):473–485. https://doi.org/10.1111/his.14792
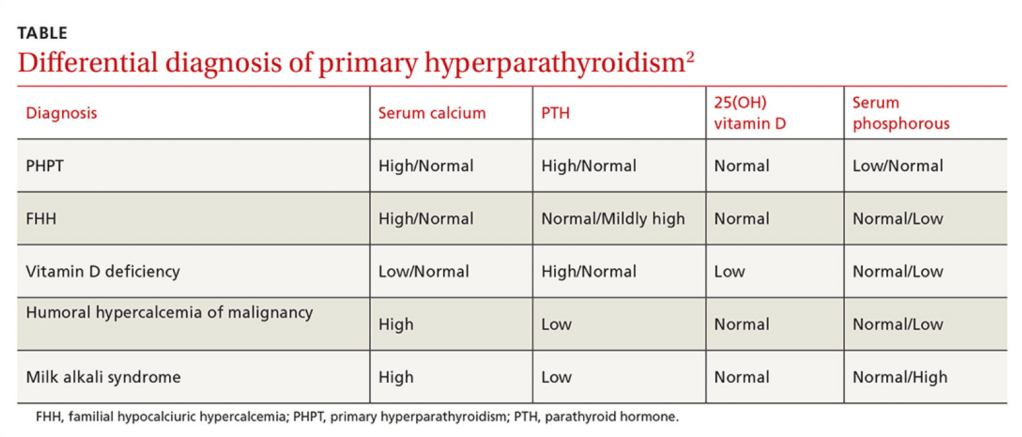
Is caused by an increased secretion of parathyroid hormone (PTH) by the parathyroid gland(s):
Which leads to an elevated serum calcium level
The overproduction of parathyroid hormone (PTH):
Termed hyperparathyroidism (HPT), can be categorized as:
Primary
Secondary
Tertiary
Primary hyperparathyroidism (PHPT);
Arises from an unregulated overproduction of PTH from an abnormal parathyroid gland
Increased PTH levels may also occur as a compensatory response to hypocalcemic states resulting from:
Chronic renal failure or gastrointestinal (GI) malabsorption of calcium:
This secondary HPT can be reversed by the correction of the underlying problem:
For example kidney transplantation for chronic renal failure
However, chronically stimulated parathyroid glands:
May occasionally become autonomous:
Resulting in the persistence or recurrence of the hypercalcemia after successful renal transplantation:
Resulting in tertiary HPT
PHPT is defined as:
Hypercalcemia or widely fluctuating levels of serum calcium resulting from:
The inappropriate or autogenous secretion of PTH:
By one or more parathyroid glands:
In the absence of a known or recognized stimulus
The most common cause of hypercalcemia in the outpatient setting is:
Primary hyperparathyroidism (PHPT):
With approximately 100,000 new cases per year reported in the United States
Since the advent of routine laboratory testing:
The prevalence of the disease has increased from:
0.1% to 0.4%:
One to seven cases per 1000 adults
In a study by Yeh et al:
The incidence of PHPT fluctuated between:
36.3 and 120.2 cases per 100,000 women-years
13.4 and 35.6 in 100,000 men-years
PHPT may present at any age:
With the vast majority of cases occurring in patients older than 45 years of age
The mean age at diagnosis has remained between:
52 and 56 years
Women have consistently made up the preponderance of cases:
With a female-to-male ratio of:
3:1 to 4:1
Based on a population based study from Rochester Minnesota:
The higher incidence of this could be secondary (hypothetically) to:
Estrogen deficiency after menopause:
That reveals underlying HPT
The precise origin of PHPT is unknown:
Although exposure to low-dose therapeutic ionizing radiation and familial predisposition account for some cases:
Irradiation for acne could have accounted for a 2 to 3-fold increase in the incidence of this disease at some point in time, and a 4-fold increase was noted in survivors of the atomic bomb
Schneider et al., in their study of 2555 patients followed for 50 years, even low doses of radiation exposure during the teenage years:
Was associated with a slight risk of developing PHPT
In this study a dose response was documented in people receiving external-beam radiotherapy for benign diseases before their 16th birthday
The latency period for the development of PHPT after radiation exposure:
Is longer than that for the development of thyroid tumors, with most cases occurring 30 to 40 years after exposure
Patients who have been radiated have similar clinical manifestations and serum calcium levels when compared to patients without a history of radiation exposure:
However, the former tend to have higher PTH levels and a higher incidence of concomitant thyroid neoplasms
Certain medications have been implicated in the development of hypercalcemia:
Lithium therapy has been known to:
Shift the set point for PTH secretion in parathyroid cells:
Thereby resulting in elevated PTH levels and mild hypercalcemia
Lithium stimulates the growth of abnormal parathyroid glands in vitro and also in susceptible patients in vivo
Unusual metabolic features associated with lithium use include:
Low urinary calcium excretion
Normal cyclic AMP excretion
Lack of calcic nephrolithiasis
The mechanism probably results from:
Lithium linking with the calcium sensing receptor on the parathyroid glands resulting in PTH secretion
Elevated serum calcium levels have been associated with thiazide diuretic:
The overall annual age- and sex-adjusted (to 2000 U.S. whites) incidence was:
7.7 (95% CI, 5.9 to 9.5) per 100,000 individuals
The average 24-hour plasma calcium concentrations are increased with thiazide diuretic use:
But the mean 24-hour PTH levels remain unchanged in subjects with normal baseline PTH levels and no evidence of hypercalciuria
Thiazides diuretics have several metabolic effects that may contribute to increased calcium levels:
A decrease in urine calcium excretion is the most likely cause:
But in some cases diuretic use has been associates with a metabolic alkalosis:
That could also increase the total serum calcium levels through a pH-dependent increase in protein-bound calcium
Although plasma 1,25 (OH) vitamin D levels are unchanged:
Increased intestinal calcium absorption in response to thiazide diurectic use:
Has been noted and could also contribute to an increase in serum calcium
One last possible explanation for the elevated serum calcium levels associated with thiazide diuretic use is:
Hemoconcentration associated with dieresis
Numerous genetic abnormalities have been identified in the development of PHPT, including:
Anomalies in tumor suppressor genes and proto-oncogenes
Specific DNA mutations in a parathyroid cell:
May confer a proliferative advantage over normal neighboring cells:
Thus allowing for clonal growth:
Large populations of these altered cells containing the same mutation within hyper functioning parathyroid tissue:
Suggest that such glands are a result of clonal expansion
The majority of PHPT cases are:
Sporadic
Nonetheless, PHPT also occurs within the spectrum of a number of inherited disorders such as:
Multiple endocrine neoplasia syndromes (MEN):
MEN type 1 (Wermer Syndrome)
MEN type 2A (Sipple Syndrome)
Isolated familial HPT
Familial HPT with jaw-tumor syndrome
All of these are inherited in an:
Autosomal dominant fashion
The earliest and most common presentation of MEN type 1 (Wermer Syndrome):
Is PHPT:
Develops in approximately 80% to 100% of patients by age 40 years
These patients also are predisposed to the development of:
Pancreatic neuroendocrine tumors
Pituitary adenomas
Less frequently:
Skin angiomas
Lipomas
Adrenocortical tumors
Neuroendocrine tumors of the:
Thymus
Bronchus
Stomach
MEN type 1 has been shown to result from:
A germline mutation in a tumor suppressor gene:
Called MEN1 gene:
Located on chromosome 11q12-13 that encodes Menin:
A protein that is postulated to interact with the transcription factors JunD and nuclear factor-κB in the nucleus, in addition to replication protein A and other proteins
Pre-symptomatic screening for mutation carriers for MEN type 1:
Is difficult because generally MEN1 mutations result in a nonfunctional proteinand are scattered throughout the translated nine exons of the gene
MEN1 mutations also have been found in kindred’s initially suspected to represent isolated familial HPT
Screening for mutation carriers for MEN type 1 has a very high detection rate greater than 94%, and is used in Sweden for patients with:
PHPT with a first-degree relative with a major endocrine tumor, age of onset is less than 30 years and / or if multiple pancreatic tumors / parathyroid hyperplasia is detected
Approximately 20% of patients with MEN type 2A (Sipple Syndrome):
Develop PHPT:
Which is usually less severe
MEN type 2A is caused by:
A germline mutation of the RET proto-oncogene:
Located on chromosome 10
Genotype and phenotype correlations have been noted in this syndrome:
In that individuals with mutations at codon 634:
Are more likely to develop PHPT
Patients with the familial HPT with jaw-tumor syndrome:
Have an increased predisposition to:
Parathyroid carcinoma
This syndrome maps to a tumor suppressor locus HRPT2 (parafibromin):
On chromosome 1
Sporadic parathyroid adenomas and some hyperplastic parathyroid glands:
Have loss of heterozygosity (LOH) at 11q13:
The site of the MEN1 gene in approximately 25% to 40% of the cases
Over expression of PRAD1:
Which encodes cyclin D1:
A cell cycle control protein:
Is found approximately 18% of parathyroid adenomas
This was proven to result from a rearrangement on chromosome 11:
That places the PRAD1 gene:
Under the control of the PTH promoter
Other chromosomal regions deleted in parathyroid adenomas and possibly reflecting loss of tumor suppressor genes include:
1p
6q
15q
Amplified regions suggesting oncogenes have been identified at:
16p
19p
RET mutations:
Are unusual in sporadic parathyroid tumors
Sporadic parathyroid cancers are characterized by:
Uniform loss of the tumor suppressor gene RB:
Which is involved in cell cycle regulation
60% have HRPT2 (CDC73) mutations located in chromosome 1:
Encodes the protein Parafibromin
These alterations are rare in benign parathyroid tumors;
May have implications for diagnosis
The p53 tumor suppressor gene:
Is also inactivated in a subset (30%) of parathyroid carcinomas
Single gland adenoma:
Is the most common cause (75% to 90%) of PHPT
Lower pole adenomas (in relation to the thyroid):
Are more common than are upper pole adenomas
Sizes range from 1 cm to 3 cm:
The normal parathyroid gland measures approximately 6 mm X 4 mm X 2 mm
The weight of parathyroid adenomas vary between:
553.7 mg +/- 520.5 mg (range, 66-2536):
The normal weight of a parathyroid gland is:
Approximately 40 mg to 50 mg
Ectopic glands can be present:
4% to 16% of cases
PHPT is caused by multiple adenomas or hyperplasia in:
15% to 25% of the cases
Parathyroid carcinoma as the cause of PHPT:
Is extremely rare in most parts of the world (~1%)
Multi-gland adenoma arises in a significant number of patients:
Double adenomas are seen in approximately:
2% to 12% of the cases
Triple adenomas:
In less than 1% the cases
Four adenomas or parathyroid gland hyperplasia:
In less than 3% to 15% of the cases
Most parathyroid adenomas:
Consist of parathyroid chief cells
They are usually encapsulated
In 50% of the cases they are surrounded by normal parathyroid tissue
Some adenomas, nevertheless, are composed of oxyphil cells:
These adenomas are usually larger than chief cell adenomas
Parathyroid adenomas are sometimes located within the thymus:
They express a parathyroid-specific gene:
GCMB
Contrasting with the normal thymus:
Which does not neither express PTH nor GCMB
In a study by Ruda et al:
225 patients with PHPT:
Parathyroid hyperplasia accounted for approximately 6% of cases
In parathyroid hyperplasia all four glands are enlarged:
With the lower glands typically being larger than the upper time
The glands are usually composed of:
Chief cells
Clear cell hyperplasia is very rare and is the only one in which the upper parathyroid glands are larger than the lower ones
👉All patients with primary hyperparathyroidism may be considered for parathyroid surgery, guidelines also include osteoporosis, kidney stones, and very high blood calcium levels as strong indications for surgery
Although exposure to low-dose therapeutic ionizing radiation and familial predisposition account for some cases
Various diets and intermittent exposure to sunshine may also be related
Other causes include:
Renal leak of calcium
Declining renal function:
With age
Alteration in the sensitivity of parathyroid gland:
To be suppression by calcium
The latency period for development of PHPT after radiation exposure:
Is longer than that for the development of thyroid tumors:
With most cases occurring 30 to 40 years after exposure
Patients who have been exposed to radiation:
Have similar clinical presentations and calcium levels when compared to patients without a history of radiation exposure:
However, the former tend to have higher PTH levels and a higher incidence of concomitant thyroid neoplasms
Lithium therapy:
Has been known to shift the set point for PTH secretion in parathyroid cells:
Thereby resulting in elevated PTH levels and mild hypercalcemia
Lithium stimulates the growth of abnormal parathyroid glands in vitro and also in susceptible patients in vivo
PHPT results from the enlargement of a single gland or parathyroid adenoma:
In approximately 80% to 95% of cases
Multiple gland disease in seen in 15% to 20% of the cases:
Doble adenomas 6% to 9% of cases:
This entity is less common in younger patients but is more prevalent in older patients
Triple adenomas < 0.3% of cases
Four gland hyperplasia 3% of cases
Parathyroid carcinoma:
In 1% of patients
It should be emphasized that when more than one abnormal parathyroid gland is identified preoperatively or intraoperatively:
The patient has hyperplasia (all glands abnormal) until proven otherwise
Genetics of PHPT:
Most cases of PHPT are sporadic:
However, PHPT also occurs within the spectrum of a number of inherited disorders such as:
MEN1 (Wermers Syndrome)
MEN2A (Sipple Syndrome)
Isolated familial HPT
Familial HPT with jaw-tumor syndrome
All of these syndromes are inherited in an autosomal dominant fashion
MEN type 1 Wermers Syndrome:
PHPT is the earliest and most common manifestation of MEN1:
It develops in 80% to 100% of patients by age 40 years old
These patients also are prone to:
Pancreatic neuroendocrine tumors:
About 50% of patients develop gastrinomas:
Which often are multiple and metastatic at diagnosis
Insulinomas develop in 10% to 15% of cases
Whereas many patients have nonfunctional pancreatic endocrine tumors
Pituitary adenomas:
Prolactinomas occur in 10% to 50% of MEN1 patients and constitute the most common pituitary lesion
Less commonly, to:
Adrenocortical tumors
Lipomas
Skin angiomas
Carcinoid tumors of the bronchus, thymus, or stomach
MEN1 has been shown to result from germline mutations in the MEN1 gene:
A tumor suppressor gene:
Located on chromosome 11q12-13:
That encodes menin:
A protein that is postulated to interact with the transcription factors JunD and nuclear factor-κB in the nucleus, in addition to replication protein A and other proteins
Most MEN1 mutations result in a nonfunctional protein and are scattered throughout the translated nine exons of the gene:
This makes presymptomatic screening for mutation carriers difficult
MEN1 mutations also have been found in kindreds initially suspected to represent isolated familial HPT
MEN type 2A Sippple Syndrome:
HPT develops in about 20% of patients with MEN2A:
It is generally is less severe
MEN2A is caused by germline mutations of the RET proto-oncogene:
Located on chromosome 10
In contrast to MEN1:
Genotype-phenotype correlations have been noted in this syndrome:
In that individuals with mutations at codon 634 are more likely to develop HPT
Patients with the familial HPT with jaw-tumor syndrome:
Have an increased predisposition to parathyroid carcinoma
This syndrome maps to a tumor suppressor locus HRPT2(CDC73 or parafibromin):
On chromosome 1
Patients belonging to isolated HPT kindreds:
Also appear to demonstrate linkage to HRPT2
Approximately 25% to 40% of sporadic parathyroid adenomas and some hyperplastic parathyroid glands:
Have loss of heterozygosity (LOH) at 11q13:
The site of the MEN1 gene
The parathyroid adenoma 1 oncogene (PRAD1):
Which encodes cyclin D1:
A cell cycle control protein:
Is overexpressed in about 18% of parathyroid adenomas
This was demonstrated to result from a rearrangement on chromosome 11 that places the PRAD1 gene:
Under the control of the PTH promoter
Other chromosomal regions deleted in parathyroid adenomas and possibly reflecting loss of tumor suppressor genes include:
1p, 6q, and 15q
RET mutations are rare in sporadic parathyroid tumors
Sporadic parathyroid cancers:
Are characterized by uniform loss of the tumor suppressor gene RB:
Which is involved in cell cycle regulation
60% have HRPT2 (CDC73) mutations
These alterations are rare in benign parathyroid tumors and may have implications for diagnosis
Whereas amplified regions suggesting oncogenes have been identified at 16p and 19p
The p53 tumor suppressor gene is also inactivated in a subset (30%) of parathyroid carcinomas